Síndrome de Angelman
SÍNDROME DE ANGELMAN
OMIM # 105830
DEFINICIÓN
El síndrome de Angelman se es un trastorno genético caracterizado por retraso severo en desarrollo, microcefalia (cabeza más pequeña de lo normal), marcha ataxica (alteraciones al caminar), ausencia de lenguaje, crisis convulsivas y comportamiento característico.
INCIDENCIA
La incidencia del síndrome de Angelman es de 1 de cada 15,000 a 25,000 recién nacidos vivos.
SINÓNIMOS
- Síndrome de la “marioneta feliz”.
CAUSA DE LA ENFERMEDAD Y CONSEJO GENÉTICO
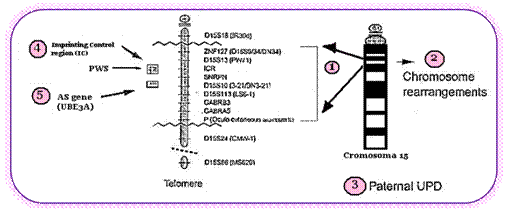
La causa del síndrome de Angelman es heterogénea, en el 70% de los casos es por deleción de un fragmento en el cromosoma 15, el riesgo de recurrencia en estos casos es <1%. Por disomía uniparental (las 2 copias del cromosoma 15 son de origen paterno, siendo lo normal tener 1 cromosoma de origen paterno y otro de origen materno) del 2-5% de los casos, el riesgo de repetición es <1%. Por mutaciones (cambios patológicos, irreversibles y heredables en la secuencia del DNA) en el gen UBE3A que se encuentra en el cromosoma 15 se explican aproximadamente 10% de los casos, si la mutación es de novo (es decir solo se presenta en la paciente) el riesgo de repetición es <1%, en caso de ser heredada el riesgo de repetición es de 50%. Por alteraciones en centro de impronta (región que controla la expresión de los genes en la región del cromosoma 15 donde dicha expresión es preferencial según origen parental) en el 2-4% de los pacientes, de estos si se trata de deleción (perdida) del centro de impronta el riesgo de recurrencia llega a ser del 50%. Finalmente en el 10-15% de los casos no se identifica la causa a nivel molecular.
CLASIFICACIÓN
No existe clasificación en el síndrome de Angelman.
MANIFESTACIONES CLÍNICAS
Las características clínicas en el síndrome de Angelman son:
- En el 100% de los pacientes:
- Retraso psicomotor severo.
- Ausencia o mínimo lenguaje.
- Marcha ataxica / temblor.
- Comportamiento característico: risa frecuente o sin motivo, movimientos estereotipados de manos, fácil excitabilidad, poca atención.
- En más del 80% de los pacientes:
- Microcefalia (que se hace evidente aproximadamente a los 2 años).
- Crisis convulsivas (comienzan antes de los 3 años).
- Electroencefalograma característico.
- En 20 a 80% de los pacientes:
- Occipucio plano.
- Atracción por el agua.
- Alteración en patrón del sueño.
- Escoliosis (desviación de la columna vertebral).
- Piezas dentales espaciadas.
- Hipopigmentación.
- Masticación y salivación excesiva.
- Protrusión lingual.
El diagnóstico del síndrome de Angelman por lo general se logra hasta los 3 a 7 años ya que las características típicas del síndrome mencionadas no se presentan hasta esta edad.
TRATAMIENTO
No hay tratamiento curativo para el síndrome de Angelman, el manejo es sintomático según se vayan presentando las manifestaciones o complicaciones por el especialista correspondiente.
Respecto a crecimiento y desarrollo el peso y la talla suelen ser normales al nacimiento, a los 6-12 meses puede presentar alteraciones en deglución y/o succión por hipotonía, así como reflujo gastro esofágico. El manejo es dietético y con ejercicio.
Para el retraso psicomotor/mental se recomienda terapia física, ocupacional, de lenguaje y de comunicación ya que la gran mayoría no se comunica de manera verbal.
Para la protusión lingual cuando es severa de puede dar tratamiento quirúrgico.
En caso de hiperactividad nos e acostumbra dar tratamiento farmacológico.
Cuando se presenta alteraciones en sueño se puede intentar dar antihistamínicos.
Se debe realizar electroencefalograma al momento del diagnóstico, las crisis convulsivas suelen controlarse con un solo medicamento. Están contraindicados los que aumenten los niveles de GABA en cerebro, como vigabatrina y carbamazepina, ya que por razones desconocidas pueden causar otro tipo de convulsiones o status epiléptico.
Para el estrabismo y alteraciones oftalmológicas se recomienda valoración por oftalmólogo cada 1 o 2 años.
Las manifestaciones musculoesqueléticas requieren valoración ortopédica, en algunos casos esta indicado el manejo quirúrgico.
NIVEL DE ATENCIÓN MÉDICA NECESARIO
- Genética
- Neurología
- Rehabilitación
- Oftalmología
- Ortopedia
COMPLICACIONES
Cuando se presentan alteraciones en deglución y/o succión pueden presentar peso y talla bajos.
En 10-15% de los pacientes no se logran controlar las crisis convulsivas con un solo medicamento, por lo cual hay que usar dos o más.
PRONÓSTICO
La expectativa de vida en los pacientes con síndrome de Angelman es casi normal, hay reportes de pacientes de hasta 74 años de edad.
Deja un comentario →


{kind=link}
{kind=link}
{kind=link}
¿Puede un niño presentar síndrome de Angelman y no tener convulsiones?
Comentar →